Recently, the Wang Tao Laboratory at the Center of Artificial Photosynthesis for Solar Fuels of Westlake University has made significant progress in the study of a new mechanism for CO methanation catalysis. Their findings were published in the Journal of the American Chemical Society. By using density functional theory (DFT) calculations and microkinetic modeling (MKM), they conducted a systematic and in-depth theoretical study on the methanation mechanism on different active centers of more than ten transition metal catalysts. They also successfully revealed the mechanism for the enhancement of catalysts through a new dual-site strategy and designed a dual-site catalyst capable of low-temperature methanation. Additionally, the systematic data generated in this work provides a high-quality database for machine-learning-based catalyst design. The first author of the paper is Zhao Wanghui, a postdoc at Westlake University, and Xu Gaomou is the co-first author, a Ph.D. student at Westlake University. Dr. Wang Tao is the corresponding author, a PI at the Department of Chemistry and the Center of Artificial Photosynthesis for Solar Fuels.
Since it was discovered by Sabatier and Senderens in 1902, the catalytic COx hydrogenation to methane (methanation) has served as an ideal model reaction for the fundamental understanding of catalysis on the gas-solid interface. This reaction plays an essential role in various industrial processes such as CH4 production, COx removal in hydrogen purification for fuel cells and ammonia synthesis processes. The conversion of CO to methane is mainly limited by challenges associated with the breaking of the strong C-O bond and thus the reaction runs at high temperatures to overcome this barrier. Like the Haber-Bosch process, the methanation reaction is an exothermic reaction and hence the increased total pressure is needed to shift to higher equilibrium conversion at elevated temperatures. Altogether, this will increase the energy consumption of the reaction, introduce higher demands on the pressure resistance of the reactor, the temperature resistance of the catalyst, and also require efficient solutions for heat transfer properties of the reactor. Therefore, developing a low-temperature solution for the methanation reaction is one of the key scientific challenges to be resolved and this clearly requires a more energy-efficient catalyst.
The research goal of Dr. Wang Tao's laboratory is to explore a stable catalyst that can dissociate CO molecules and hydrogenate CHx and O species at a lower energy barrier. Based on the previous work of Dr. Wang Tao's group in the synthesis of ammonia under mild conditions, they proposed a design strategy for effectively activating CO with dual-site catalysts to break through this bottleneck (Figure 1).
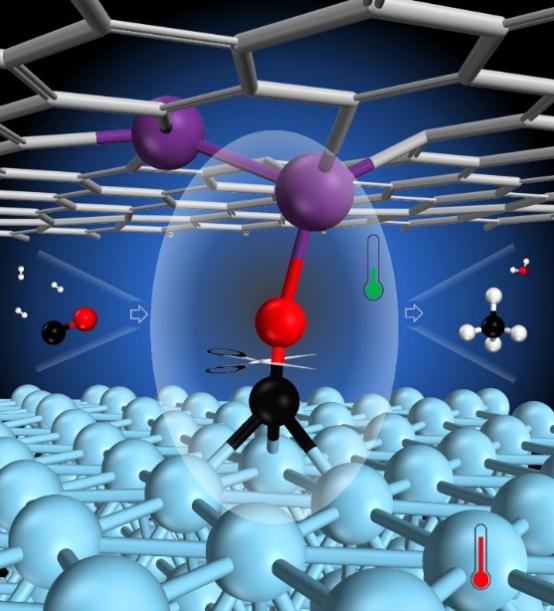
Figure 1 Schematic diagram of the structure of an ideal dual-site catalyst
Due to the complexity of the CO hydrogenation reaction mechanism, mechanistic studies in the field of theoretical catalysis have mainly focused on individual metal catalysts. To have a more comprehensive and in-depth understanding of this complex mechanism, Dr. Wang Tao's team systematically calculated the CO hydrogenation reaction mechanism of more than ten transition metal catalysts with step sites and dual sides. As shown in Figure 2A, the dissociation energy barrier of CO on dual-side metal catalysts is significantly lower than that of step sites, which preliminarily demonstrates the superiority of dual-site catalysts in CO dissociation. When combined with a mean-field MKM, a volcano-shaped relationship between the calculated TOF of methane production (TOFCH4) and ΔEC_O can be plotted (Figure 2B), which shows how reactive metals are limited by the hydrogenation of surface species whereas less reactive metals are limited by the breaking of the strong C-O bond as represented by the Sabatier principle. Evidently, Co and Ni with moderate C and O binding strengths are very close to the top of the activity volcano, displaying 2 and 4 orders of magnitude higher TOFCH4 than their stepped model surfaces, respectively, suggesting that the strategy of dual active sites to activate inert chemical bonds is feasible.
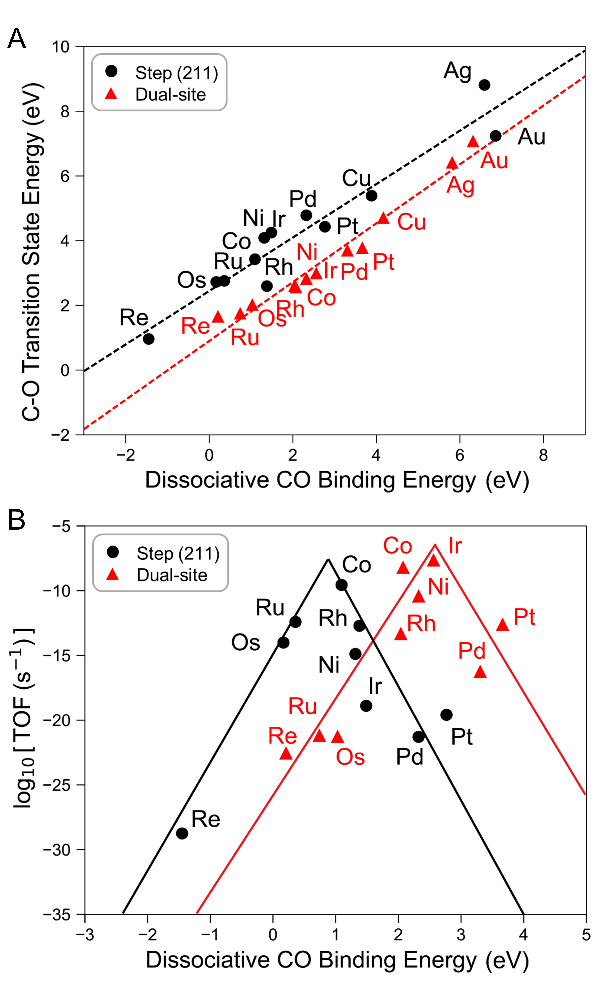
Figure 2 CO dissociation energy barrier on metal catalysts (A) and the relationship between TOFCH4 and dissociative CO binding energy at 523 K / 3 bar (B).
However, there is still enough space to further optimize the Co-based dual-site catalyst to reach the peak of the volcano. In-depth analysis reveals that the Co dual-site catalyst is on the left side of the volcano, and its activity is mainly limited by the hydrogenation step of O and OH species. Therefore, if we could identify an active center (AC) with a slightly weaker O binding that simultaneously retains the capability of breaking the C-O bond with a low energy barrier, then such an integrated Co-AC dual-site system should result in a further increase in CO methanation activity. Finally, a novel Co-Cr2/G dual-site catalyst shown in Figure 3C was designed based on theoretical calculations. At the same reaction conditions, the Co-Cr2/G catalyst show 4-6 orders of magnitude higher TOFCH4 than the stepped Co surface, as shown in Figure 3F. This study proposes a theoretical mechanism for the efficient activation of inert small molecules, further validates the feasibility of the dual-site strategy, and demonstrates the potential to achieve methanation under mild conditions.
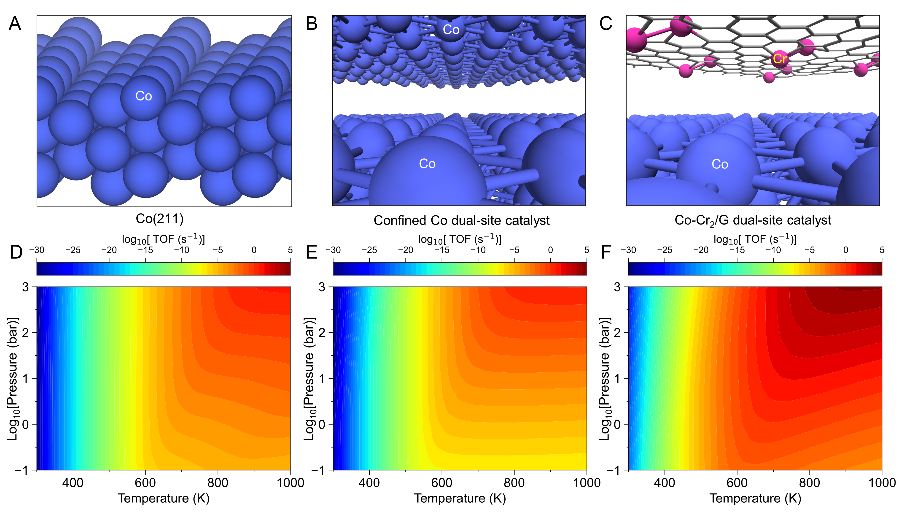
Figure 3 Schematic structures of stepped Co(211) surface (A), confined Co dual-site catalyst (B), and Co-Cr2/G dual-site catalyst (C). The calculated TOF of CH4 as a function of temperature and total pressure (pCO:pH2 = 1:2) on the Co(211) surface (D), the Co dual-site catalyst (E), and the Co-Cr2/G dual-site catalyst (F), respectively.
This work was supported by the special funds of WestLake University, the National Natural Science Foundation of China, the Research Center for Industries of the Future (RCIF) at Westlake University, and the HPC Center of Westlake University.