孙立成院士团队最新Nature Catalysis
浏览量:时间:2022年05月26日 10:43

背景介绍
析氧反应(OER)是一种理想的阳极反应,为制氢、CO2和N2还原以及其他电化学反应提供电子和质子。然而,OER受到两个水分子中四个电子的复杂去除、O-H键断裂和O-O键形成的阻碍。因此,开发高效、稳定的OER催化剂是将可再生能源转化为化学燃料的首要挑战,需要对催化过程进行全面的机理理解。人们一直致力于探索过渡金属基OER催化剂。由于结构和电子性质的易调性,以及介导多个质子-电子转移反应的能力,具有特定结构的分子水氧化催化剂(WOC)受到了广泛关注。然而,它们的不稳定性以及繁琐的合成和固定化过程限制了它们的广泛应用。相比之下,使用更耐用的过渡金属基多相催化剂缺乏对水氧化机理和结构-活性关系的全面阐述。最近,能够锚定孤立单一金属位点的各种负载材料的开发取得了巨大进展。然而,大多数用于固定单个金属位点的负载材料是不均匀的,这导致金属原子被不同的化学环境包围。这些催化位点的整体异质性限制了它们在催化机理研究中的应用。因此,一个能够提供明确配位环境以形成结构明确的过渡金属活性位点的催化系统对于非均相OER的可靠机理研究至关重要。
本文亮点
1. 本文报道了锚定在Aza CMP材料上单中心Co2+作为OER的活性中心,合成的Aza CMP Co在中性和碱性条件下都能高效催化OER,其TOFs高于最先进的材料催化剂。
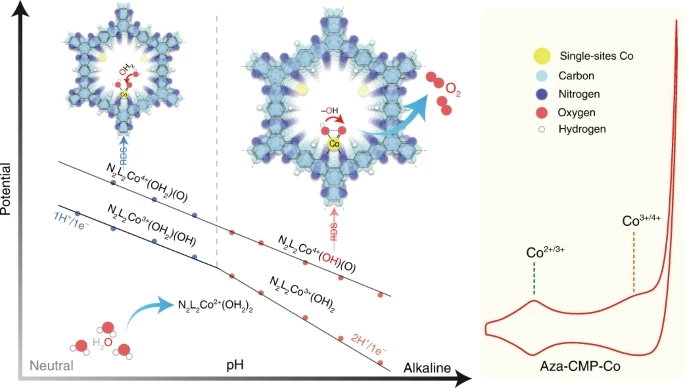
2. 结合实验结果和理论结果,提出了一种pH依赖的O-O键形成亲核攻击途径。在碱性条件下,邻羟基亲核攻击Co4+=O的分子内羟基亲核攻击(IHNA)过程被确定为速率决定步骤。与分子间水亲核攻击(WNA)途径相比,该过程导致更低的活化能和加速的动力学。
图文解析
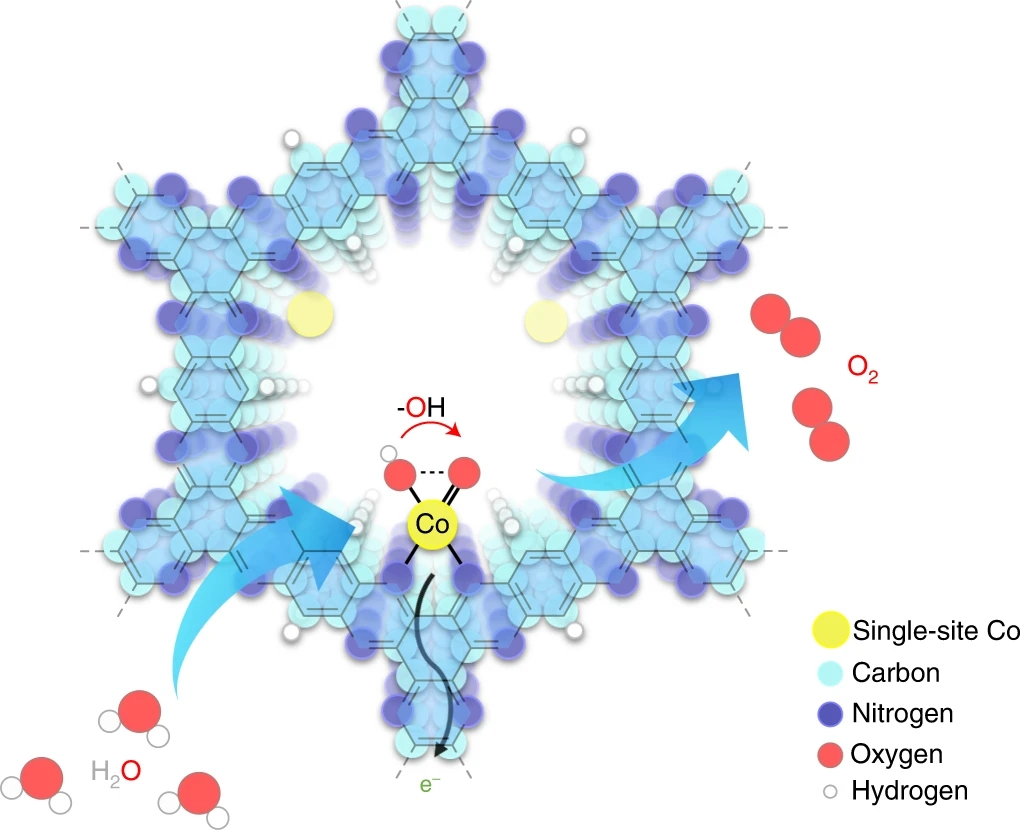
图1. 碱性条件下Aza-CMP-Co上的析氧反应示意图
要点: Aza-CMP由1,2,4,5-benzenetetramine tetrahydrochloride和triquinoyloctahydrate缩聚合成,含有许多分散良好的周期性吡啶氮配位位点。与包含各种结合模式的二维石墨烯基非均相单原子催化剂相比,Aza-CMP仅包含一种吡啶氮。随后与Co2+配位形成Aza-CMP-Co,用于水氧化。
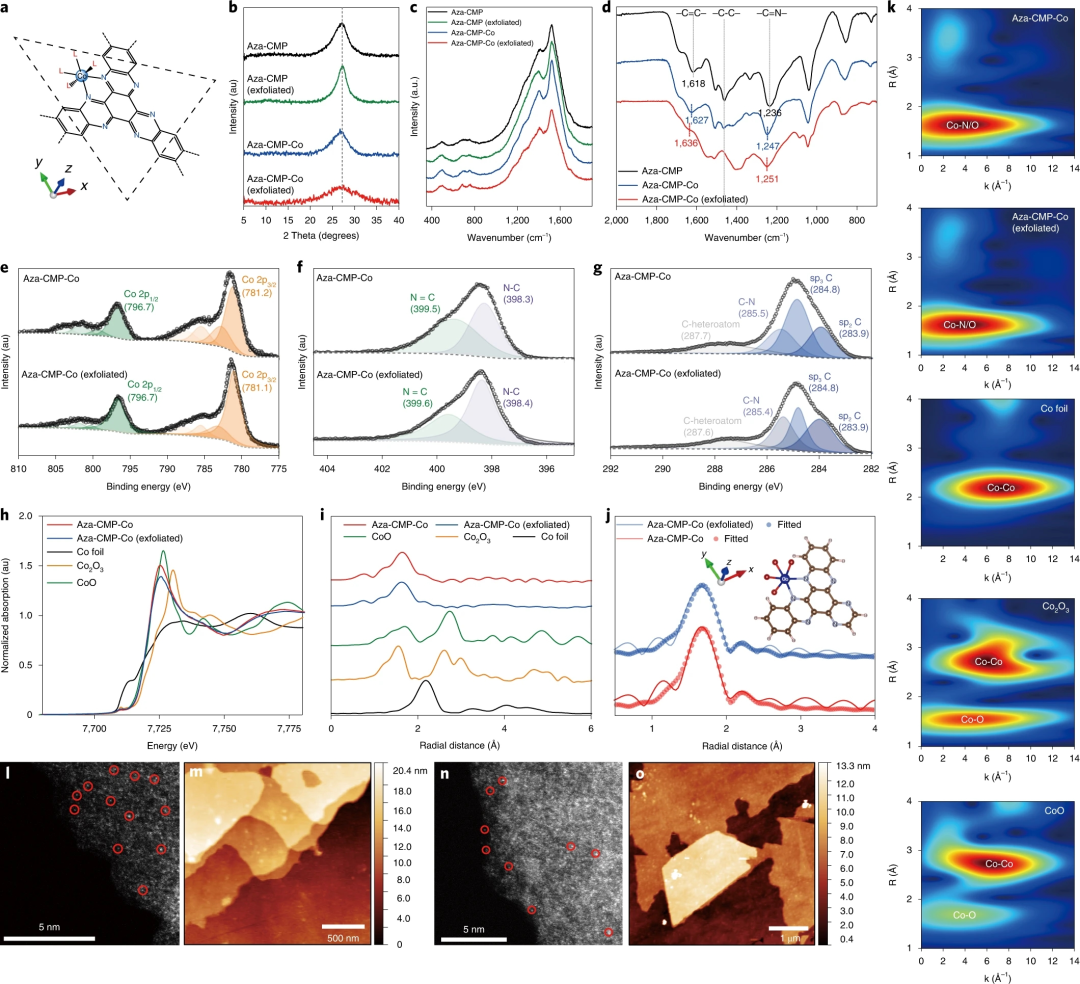
图2. Aza-CMP-Co催化剂的组成、结构和形态表征
要点1:电感耦合等离子体-光学发射光谱 (ICP-OES)显示,制成的Aza-CMP-Co的总Co2+含量为1.86 wt%,远低于 Aza-CMP-Co 的理论最大负载量。在37% HCl中液相质子化辅助剥离Aza-CMP后,合成的Aza-CMP-Co (exfoliated) 的负载量(19.2 wt%)比未经处理的Aza-CMP-Co负载量高10倍。
要点2:Aza-CMP-Co和Aza-CMP-Co (exfoliated) 的粉末X射线衍射图谱与原始Aza-CMP和Aza-CMP-Co (exfoliated) 的一致,表明层状结构在Co2+固定后保持不变。通过X射线光电子能谱也可以发现,超声后材料的键连方式和Co2+的配位环境没有发生明显变化。
要点3:对于Aza-CMP-Co、Aza-CMP-Co(exfoliated)、Aza-CMP 和 Aza-CMP (exfoliated) 获得的拉曼光谱证明在络合过程中没有产生氧化钴相。在FT-IR光谱中,C-C、C=C 和C=N的特征峰在Co2+结合后仍然存在,证明Aza-CMP骨架的化学结构在Co2+改性后得到了很好的保留。复合后观察到 ν(C=N) 的蓝移,当更多的Co2+离子锚定在N位点时,Aza-CMP-Co(exfoliated) 的 ν(C=N) 进一步移动到1251 cm-1,对于-C=C-振动,观察到类似的蓝移趋势。此外,随着钴负载量的增加,-C-C-振动的强度降低,表明局部配位环境发生了变化。
要点4:Aza-CMP-Co和Aza-CMP-Co (exfoliated) 的XANES曲线几乎相同,这表明两种催化剂采用了相同的配位环境。制备的催化剂的归一化CoK边吸收阈值与CoO相似,表明钴的价态约为+2。
要点5:Aza-CMP-Co和Aza-CMP-Co (exfoliated) 的FT-EXAFS光谱在第一壳层区域具有几乎相同的主峰,表明相同的配位构型。第二壳层区域没有峰,表明没有钴基金属或氧化物相,这进一步证实了Aza-CMP-Co催化剂中单中心钴离子的形成。
要点6:即使钴负载量达到20wt%,Aza-CMP-Co中的钴离子以单核金属中心的形式存在。
要点7:催化剂膜的AFM图像中可以观察到厚度约为4 nm的层状结构,这意味着层状CMP基质在Co2+络合后保持其结构。单一金属元素在原子分辨率的HAADF-STEM图像中很容易看到。图2l中的亮点表明Co原子分散在Aza-CMP-Co中而没有聚集的纳米颗粒。图2n中的高密度亮点表明剥离催化剂含有比Aza-CMP-Co更多的钴位点,这与ICP-OES结果一致。
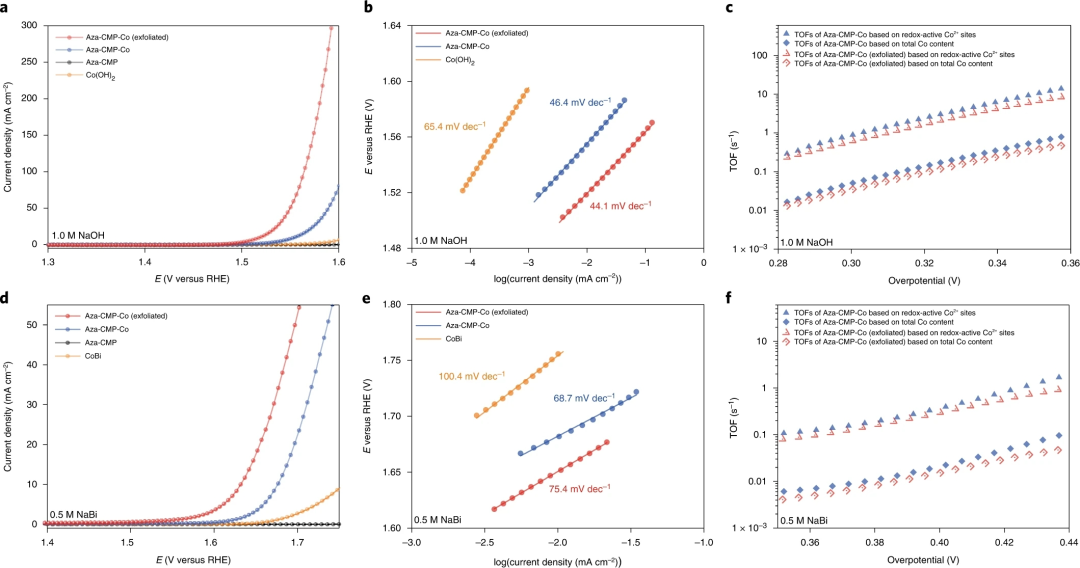
图3. Aza-CMP-Co催化剂的OER催化性能
要点1:将典型的Co(OH)2催化剂电沉积在CP基底上,与Aza-CMP-Co进行直接比较电催化OER性能,并将原始的Aza-CMP/CP样品作为对照。极化曲线显示Aza-CMP-Co (exfoliated) /CP表现出最高的催化活性。
要点2:Aza-CMP-Co(exfoliated)/CP和 Aza-CMP-Co/CP的起始电位分别为~1.47和1.48 V(0.1 mAcm-2时与可逆氢电极RHE相比的电位)。相比之下,Co(OH)2/CP电极的起始电位为1.58 V。Aza-CMP-Co (exfoliated)/CP和Aza-CMP-Co/CP表现出相似的Tafel斜率,约为44 mVdec-1,而电沉积Co(OH)2/CP表现出更高的Tafel斜率,为65.4 mVdec-1,这意味着Aza-CMP-Co (exfoliated)和Aza-CMP-Co经历了相同的OER反应途径,与Co(OH)2的反应途径显著不同。
要点3:得益于快速催化动力学,Aza-CMP-Co (exfoliated)/CP电极在电流密度为10和100 mAcm-2时的过电位要求分别仅为289和335 mV。由于金属负载水平相对较低,Aza-CMP-Co/CP电极需要324和370 mV的过电位才能分别达到相同的电流密度。
要点4:Aza-CMP-Co在整个电位范围内表现出最好的TOF,在300 mV的过电位下达到 0.95 s-1的TOFredox-active,在350mV的过电位下进一步达到10.6 s-1。对于剥离样品,在300和350 mV的过电位下,获得了较低的TOFredox-active值,分别为0.73和7.7 s-1。此外,Aza-CMP-Co和 Aza-CMP-Co (exfoliated) 的TOFtotal-content分别在313和321 mV的过电位下达到 0.1 s-1,这些结果优于大多数用于电催化碱性水氧化的氧化钴/氢氧化钴基催化剂。
要点5:在近中性条件下(0.5 M 硼酸钠缓冲液,pH 9.2)进一步研究了Aza-CMP-Co的OER 性能。Aza-CMP-Co (exfoliated)催化剂需要309和400 mV的过电位才能分别达到1和10 mAcm-2的电流密度,相应的塔菲尔斜率为 75.4 mVdec-1,表明在近中性条件下相对快速的 OER 动力学。对于Aza-CMP-Co/CP电极,电流密度为1和10 mAcm-2时的过电位分别为388和446 mV。
要点6:在近中性条件下,Aza-CMP-Co (exfoliated)/CP和Aza-CMP-Co/CP的TOFredox-active和 TOFtotal-content值在360和440mV之间的过电位下呈指数增加,Aza-CMP-Co/CP的TOFredox-active在425mV的外加过电位下达到每个氧化还原活性Co2+位点1.0 s-1,并在450 mV的过电位下进一步增加到2.9 s-1,而剥离样品在439 mV的过电位下为1.0 s−1。
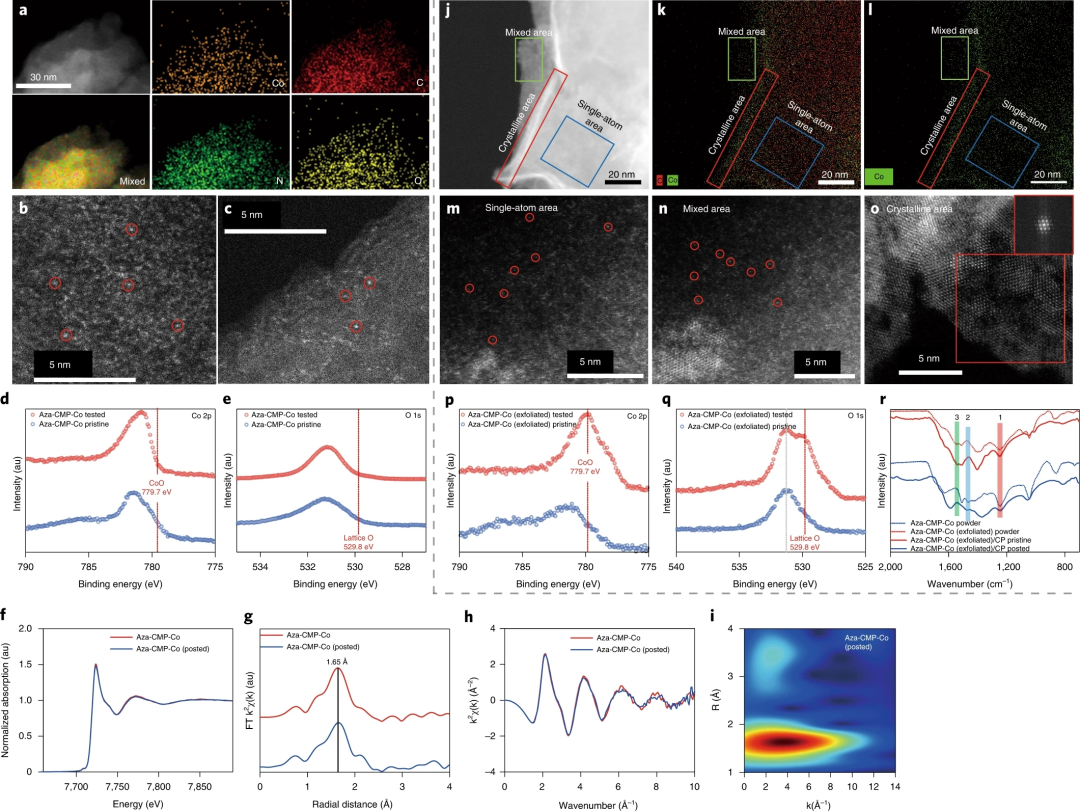
图4. 测试的Aza-CMP-Co催化剂的组成、结构和形态表征
要点1:EDX元素映射证实了OER后Aza-CMP-Co中钴位点的均匀分布,HAADF-STEM 图像表明Aza-CMP框架中的钴原子保持原子分散而没有聚集。样品较薄边缘的STEM图像显示了 Aza-CMP 的层状结构,并且单金属位点的存在很明显。使用XPS分析OER后Aza-CMP-Co的组成和电子态,Aza-CMP-Co的高分辨率光谱对应于原始样品的光谱,在Co2p 和O1s区域中不存在氧化物相。
要点2:图4f-h中几乎一致的曲线表明OER后钴位点周围的第一配位壳的变化很小。此外,Aza-CMP-Co和Aza-CMP-Co (posted) 的WT分析结果分别在3.90和 3.87 Å-1处表现出相似的强度最大值,表明Aza-CMP-Co在OER后依然存在着单核金属中心。
要点3:对于Aza-CMP-Co (exfoliated),在EDX元素映射图像中观察到钴组分的聚集,图 4m显示了催化后选定区域中存在的大量钴位点。然而,在OER期间也产生了纳米氧化物簇和结晶氧化物切片。与催化前Aza-CMP-Co (exfoliated)的XPS光谱相比,779.7 eV处的Co 2p峰偏移和529.8 eV处的新O 1s峰证实了氧化钴物种的存在。
要点4:Aza-CMP中钴成分的动态演变随金属位点负载而变化:在金属负载量相对较低的情况下,Aza-CMP-Co表现出优异的结构稳定性,而钴位点负载量增加到约20 wt%,由于受限局部区域中高浓度的分子钴导致钴位点的明显分解。
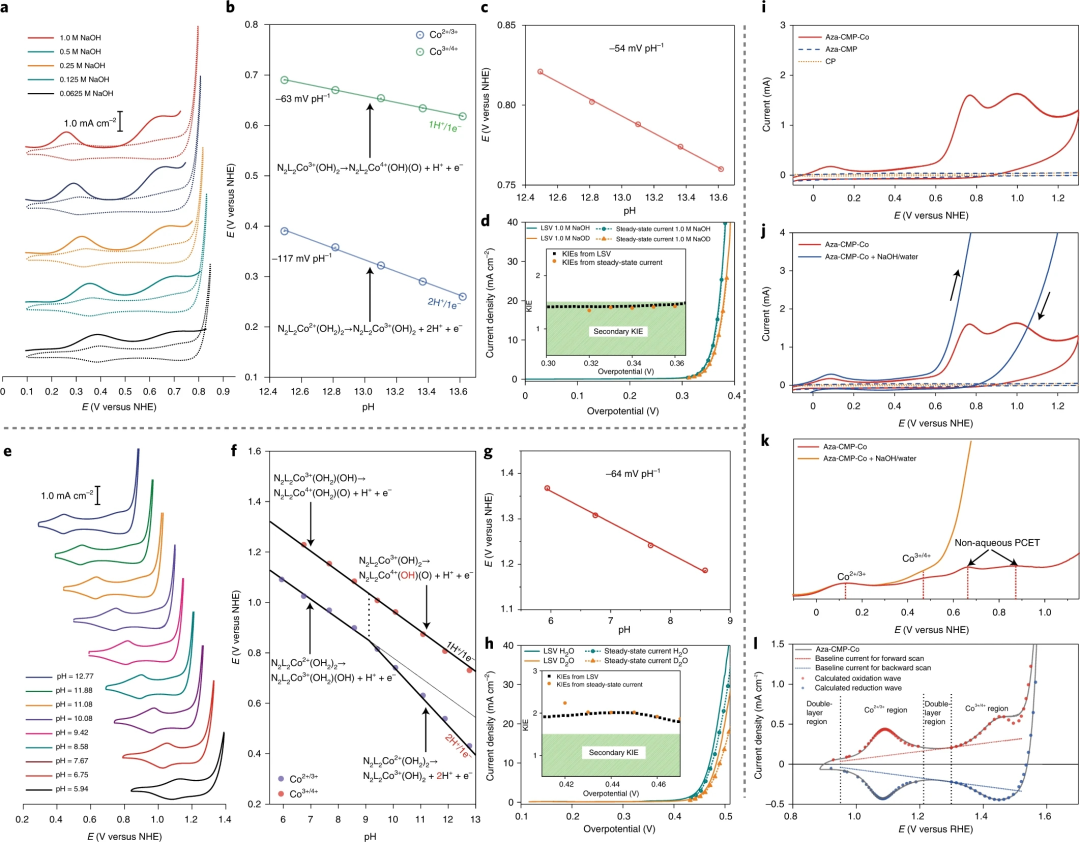
图5. Aza-CMP-Co催化剂的pH依赖性、KIEs和氧化还原态研究
要点1:如 Pourbaix 图所示,Co2+/3+氧化还原的斜率为-117 mVpH-1,对应于双质子/单电子转移过程(2H+/1e-)。对于Co3+/4+氧化还原对,斜率为-63 mVpH-1,对应于1H+/1e-转移过程。
要点2:如图f所示,绘制在pH6.0-13.0范围内的氧化还原电位与缓冲阴离子的类型无关,在pH6.0–9.0范围内观察到的第一个氧化还原过程被指定为[N2L2Co2+(OH2)2]氧化为 [N2L2Co3+(OH2)(OH)],同时损失一个质子。在pH>9.0时,Co2+/3+线出现了一个明显的拐点,根据-115 mVpH-1的斜率可认为是 [N2L2Co2+(OH2)2]到[N2L2Co3+(OH)2]的双质子转移过程。
要点3:与1.0 M NaOH水溶液相比,Aza-CMP-Co/CP在1.0M NaOD D2O溶液中的LSV 显示出较低的电流密度。在0.30–0.36 V的催化过电位范围内,相应的KIEH/D值 <1.5。在碱性条件下,在0.30–0.36V的过电位范围内,计时电流法测量的KIE值接近1.4。用不同比例的氘(n)进一步检查碱性条件下的KIE值。随着OD-浓度增加,KIE不断增加但始终保持<1.5,表明O-H键断裂可能不直接参与RDS的二级KIE。
要点4:对于近中性条件,可以在硼酸盐缓冲液中在催化过电位范围内确定较大的KIEH/D 值(>1.8),证明主要的KIE效应。Aza-CMP-Co中不存在主要的KIE,这表明在碱性条件下,(1) O-H键断裂不参与RDS,(2) 解耦的ET过程可能是OER的RDS。
要点5:Aza-CMP-Co表现出可忽略不计的pH依赖性OER活性,Co2+/3+氧化还原的峰值电流在不同的pH条件下保持一致,表明Aza-CMP-Co中的反应活性位点的数量对酸碱度变化不敏感。这揭示了Aza-CMP-Co催化OER是一种典型的非均相催化反应,其反应速率取决于催化剂上活性位点的数量和催化驱动力,而不取决于反应物浓度。
要点6:pH非依赖性活性和初级KIE满足近中性条件下的Aza-CMP-Co协同APT机制的行为,然而缺乏主要KIE表明,在碱性条件下Aza-CMP-Co发生了非协同质子-电子转移RDS。
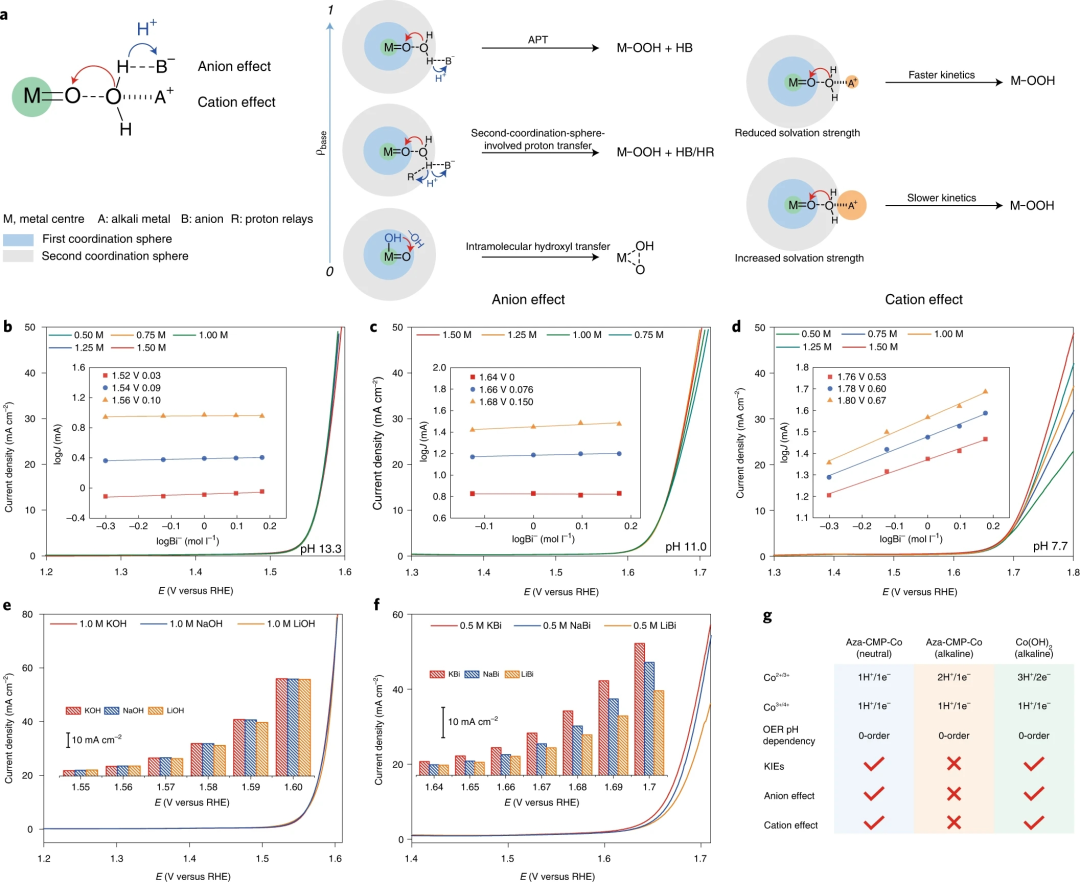
图6. 外球面环境影响的电化学研究
要点1:扩散控制的溶液APT过程受电解质中外部阴离子浓度的影响,当OER的 RDS通过APT途径进行时,在催化剂修饰的电极中广泛观察到与阴离子有关的一级反应。对于硼酸盐缓冲液,阴离子效应在 pH 11.0 和13.3时不存在,但在 pH 7.7时明显。
要点2:对于硼酸盐缓冲液中的OER,碱性阳离子K+和Li+之间的活性降低,表明氢键网络和溶剂化强度对溶液APT过程有显著影响。然而,Aza-CMP-Co在碱性pH下的催化性能与阳离子物种无关。
要点3:Aza-CMP-Co的OER活性在近中性条件下清楚地显示出对阴离子浓度和阳离子种类的依赖性,表明溶液APT过程是RDS。而在碱性条件下没有观察到这种依赖性,这意味着RDS不受额外质子受体和不同溶剂化强度的影响。在碱性条件下Aza-CMP-Co不存在 KIE,这表明直接O-H键断裂不参与RDS。
要点4:碱性条件下不存在溶剂化强度依赖性是由RDS中的分子内羟基转移过程引起的。
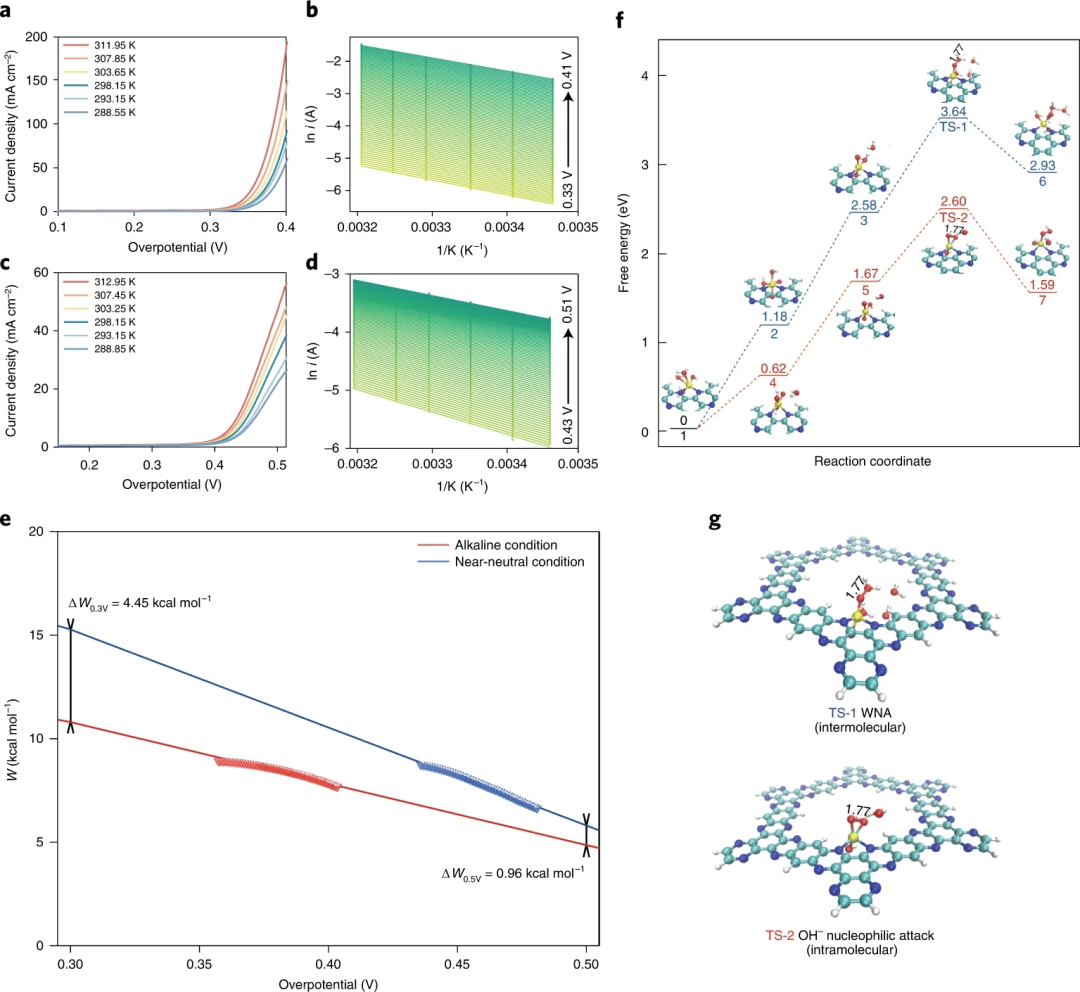
图7. 实验和理论反应活化能
要点1:Aza-CMP-Co的OER活性在NaOH和NaBi溶液中都表现出精确的温度依赖性特征,碱性条件下的W值(Walkaline)显著低于近中性条件下的W值(Wneutral)。由于活化能与外部因素无关,因此观察到的W差异表明不同的RDS中间状态,这可能导致内在性能的显著差异。
要点2:在碱性和近中性电解质中催化的OER性能之间的巨大性能差距的关键是pKa诱导的、pH相关的亲核攻击途径。
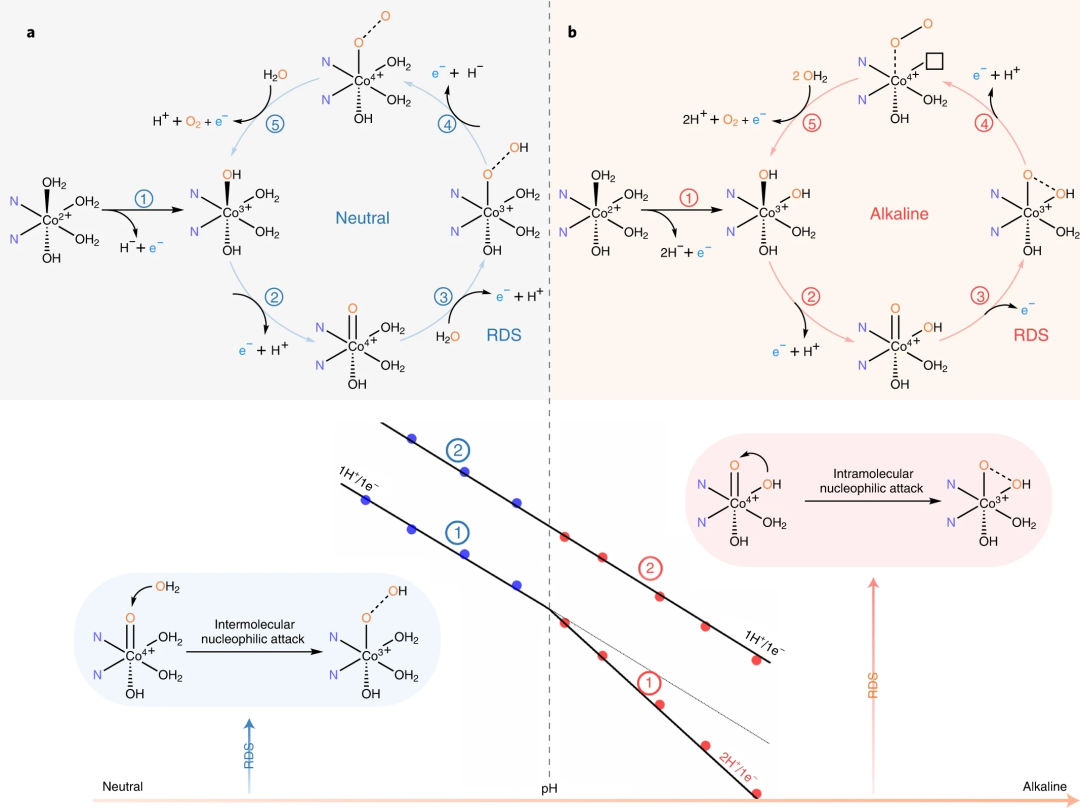
图8. 提出的OER机制
要点1:在Co2+向Co3+的转化过程中发生去质子化,当pH值增加到高于Co3+配合物的pKa 值时,两个质子分离。根据 Pourbaix 图,当Co3+进一步氧化为Co4+时,一个OH-配体被去质子化,在近中性和碱性条件下形成Co4+=O 片段。当反应在缓冲介质中进行时,缓冲液的阴离子组分可以作为溶液APT机制中的质子受体。在碱性 pH 范围内,协同质子-电子氧化涉及OH-作为质子受体发生。在非水溶液中Co3+/Co4+电位后至少观察到两个连续的氧化还原过程,当OER发生时,这些氧化还原特征立即减弱。因此,RDS步骤3 是O-O键形成过程,涉及协同溶液APT过程,在近中性条件下形成Co-OOH片段。然而,实验证据和计算结果表明,在不同的pH值下,O-O的形成机制存在显著差异。由于在碱性电解质中Co2+/Co3+的非协同PCET过程中形成额外的-OH基团,CoOOH片段倾向于通过分子内羟基亲核攻击途径生成,其中相邻的-OH攻击Co4+=O中的氧配体,形成分子内O-O键,在RHE尺度上产生二级KIEH/D和pH无关的OER活性。在中性条件下,该途径需要比分子间WNA低得多的活化能。最后,-OOH配体在进一步的1H+/1e- PCET过程后转化为分子氧,并释放O2并重新附着底物以完成催化循环。
要点2:与IHNA途径相比,具有更高表观活化能的分子间WNA途径在中性条件下占主导地位并阻碍了快速 OER 动力学。
全文总结
本文报道了锚定在Aza CMP材料上单中心Co2+作为OER的活性中心,合成的Aza CMP Co在中性和碱性条件下都能高效催化OER,其TOFs高于最先进的材料基催化剂。结合实验结果和理论结果,提出了一种pH依赖的O-O键形成亲核攻击途径。这项研究对电解质pH在水氧化催化中的关键作用以及通过调节IHNA途径增强水氧化活性提供了重要的见解。
全文信息
Intramolecular hydroxyl nucleophilic attack pathway by a polymeric water oxidation catalyst with single cobalt sites
H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, I. Furó, M. SG Ahlquist, L. Sun
Nat. Catal. 2022, 5, 414-429.
全文链接:https://www.nature.com/articles/s41929-022-00783-6
本文经授权转载自"研之成理 "
来源:研之成理
原文链接:https://mp.weixin.qq.com/s/vcUb556jT_u-mxuAuAAy7w